Pulmonary hypertension
Notes
Overview
Pulmonary hypertension refers to an increase in the pressure of the pulmonary arteries.
Pulmonary Hypertension (PH) refers to increased pressure in the pulmonary arteries due to any cause. Increased pulmonary arterial pressure is problematic because, over time, the right ventricle has to work harder to pump blood around the pulmonary circulation. Initially, the right ventricle will hypertrophy to cope with the increased pressure. However, if pulmonary artery pressure remains high, the right ventricle will eventually dilate and fail, leading to right heart failure.
Pulmonary Arterial Hypertension (PAH) is a specific type of Pulmonary Hypertension, caused by increased resistance in small pulmonary arterioles.
Definitions & physiology
Pulmonary hypertension is generally defined as a mean pulmonary arterial pressure ≥20 mmHg.
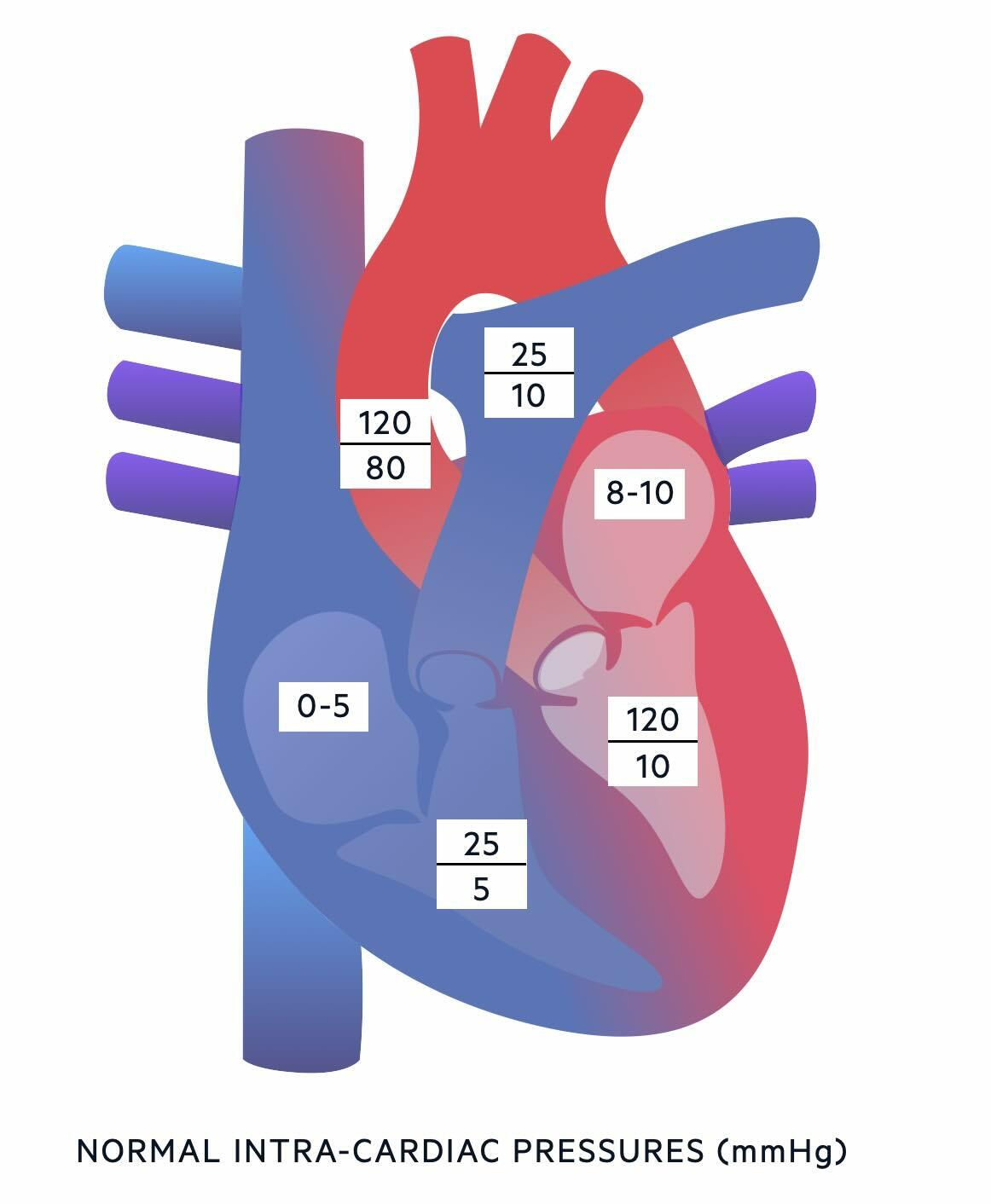
Intracardiac pressures
To understand PH, you need to know the normal pressures within the different chambers of the heart and great vessels:
- Right atrium: 0-5 mmHg
- Right ventricle: 25 mmHg / 5 mmHg
- Pulmonary artery: 25 mmHg / 10 mmHg
- Left atrium: 8-10 mmHg
- Left ventricle: 120 mmHg / 10 mmHg
- Aorta: 120 mmHg / 80 mmHg
Definitions
- Systolic Pulmonary Artery Pressure (PAP) =
- the maximal blood pressure in the pulmonary artery over the cardiac cycle. A normal systolic PAP is roughly 18 - 25 mmHg.
- Mean Pulmonary Artery Pressure (PAP) =
- the average blood pressure in the pulmonary artery over the cardiac cycle (calculated similarly to mean systemic blood pressure):
- Mean PAP = 2/3 Diastolic PAP + 1/3 Systolic PAP
- A normal mean pulmonary artery pressure is around 11-17mmHg.
- the average blood pressure in the pulmonary artery over the cardiac cycle (calculated similarly to mean systemic blood pressure):
- Pulmonary Hypertension (PH) =
- a mean pulmonary artery pressure greater than 20mmHg at rest*. Pulmonary hypertension is an umbrella term that includes 5 ‘groups’ of different causes.
*PH used to be defined as mPAP ≥ 25mmHg, and you may still see this value used. Following the 6th World Symposium on Pulmonary Hypertension in 2019, there is a move towards lowering the cut-off for PH to a mPAP ≥ 20mmHg.
- Pulmonary Arterial Hypertension (PAH) =
- a specific form of pulmonary hypertension characterised by increased resistance to blood flow in small pulmonary arterioles. Pulmonary hypertension in the absence of heart or lung disease, chronic thromboembolic disease, or other rare causes. Also known as:
- Group 1 pulmonary hypertension (see ‘classification’ below)
- Idiopathic pulmonary arterial hypertension
- a specific form of pulmonary hypertension characterised by increased resistance to blood flow in small pulmonary arterioles. Pulmonary hypertension in the absence of heart or lung disease, chronic thromboembolic disease, or other rare causes. Also known as:
- Cor Pulmonale =
- right-sided heart failure due to a primary problem in the lungs and associated pulmonary hypertension. Also known as:
- Group 3 pulmonary hypertension (see “classification” below).
- right-sided heart failure due to a primary problem in the lungs and associated pulmonary hypertension. Also known as:
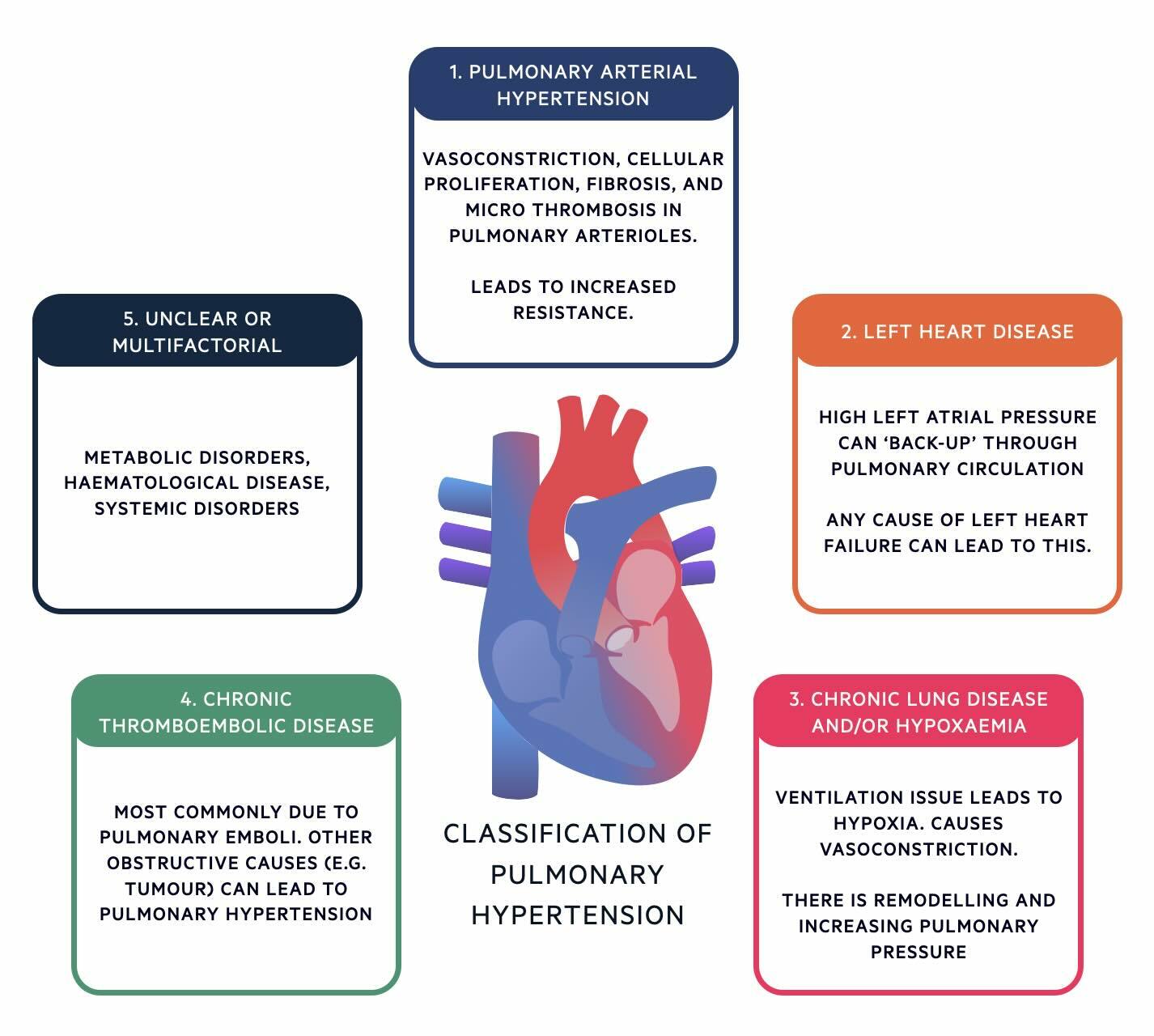
Classification
Pulmonary Hypertension is divided into 5 groups based on its underlying cause.
The classification of pulmonary hypertension can be remembered using the mnemonic 'PLATE':
- P (group 1): Pulmonary artery hypertension
- L (group 2): Left heart disease
- A (group 2): Airway disease (chronic lung disease and/or hypoxaemia)
- T (group 4): Thromboembolic disease (or other obstructive conditions)
- E (group 5): Everything else (anything unclear or multifactorial)
Epidemiology
Pulmonary Hypertension (of any cause) is a relatively rare condition.
Pulmonary Hypertension (of any cause) is relatively rare: 15 – 50 people per million are affected by it i.e. 0.0015 – 0.005%.
In the UK, the most common causes of Pulmonary Hypertension are:
- Group 2: PH due to Left Heart Disease, and
- Group 3: PH due to Chronic Lung Disease and/or Hypoxemia.
Pulmonary arterial hypertension is rare (group 1). It affects 5 to 15 people per million. It usually presents in patients in their 40s and 50s and is roughly twice as likely to affect women as compared to men. PH due to Pulmonary Artery Obstruction (Group 4) develops in 0.5 - 4% of patients after acute pulmonary embolism.
Aetiology & pathophysiology
The pathophysiology of pulmonary hypertension differs depending on the underlying cause.
Group 1: PH due to Pulmonary Arterial Hypertension
The final pathophysiological change in Group 1 PH is the remodelling of small pulmonary muscular arterioles (i.e. arterioles < 50 microns). All 3 layers of the vascular wall (i.e. intima, media, and adventitia) undergo changes, which include vasoconstriction, hyperplasia, hypertrophy, fibrosis, and thrombosis. The consequence of these changes is an increased resistance to blood flow, leading to pulmonary hypertension.
The following underlying causes can lead to this remodelling:
- Idiopathic PAH (a cause is not identified, and there is no family history of PAH).
- Heritable PAH (a specific genetic mutation leading to remodelling is identified). Examples of these include BMPR2, ALK1 and 5HTT.
- Drugs and toxins (e.g. methamphetamines, cocaine)
- HIV infection
- Connective Tissue Disease (e.g. systemic sclerosis, systemic lupus erythematosus, rheumatoid arthritis).
NOTE: Connective tissue disease is complex and may cause pulmonary hypertension via various mechanisms not simply restricted to one group.
Group 2: PH due to Left Heart Disease
Any cause of increased left atrial pressure can lead to backpressure through the pulmonary circulation.
The commonest causes of increased left atrial pressures are:
- HFrEF: Left-sided heart failure with reduced ejection fraction.
- HFpEF: Left-sided heart failure with preserved ejection fraction.
- Mitral stenosis.
- Aortic stenosis.
Group 3: PH due to Chronic Lung Disease and/or Hypoxemia
The final pathophysiological change in Group 3 PH is irreversible hypoxic vasoconstriction.
It may help your understanding to recall that pulmonary blood vessels respond differently to systemic blood vessels in response to hypoxia:
- Systemic blood vessels (e.g. in muscles) dilate in response to hypoxia. This helps direct blood towards metabolically active tissue where oxygen is needed most.
- Pulmonary blood vessels constrict in response to hypoxia. This helps direct blood away from areas of the lung with little oxygen, and towards areas with plenty of oxygen, maximising the amount of oxygen blood collects on its passage through the lungs.
In the lungs, this process is also known as ventilation/perfusion matching.
- Short-term hypoxic pulmonary vasoconstriction is a useful physiological mechanism that matches perfusion with ventilation. It is a reversible process.
- Long-term hypoxic pulmonary vasoconstriction is problematic. It can lead to more permanent, irreversible changes to the pulmonary vasculature.
We can split the causes of Group 3 PH into two categories:
- Chronic lung disease (e.g. COPD and Interstitial lung disease)
- Hypoventilation (e.g. Obstructive sleep apnoea)
Group 4: PH due to Pulmonary Artery Obstruction
The commonest cause of Group 4 PH is pulmonary emboli.
After a pulmonary embolism and treatment with anticoagulation, most patients will fully resorb the blood clot, and normal pulmonary blood flow through the lungs will resume.
In a small subset of patients, following a pulmonary embolism and treatment with anticoagulation, resorption of the blood clots is incomplete. Instead, the clot becomes ‘organised’ – it changes from fresh red to yellow in colour, fills with collagen and elastin, and becomes firmly adherent to the walls of the pulmonary vessels. These persistent blood clots obstruct blood flow through the pulmonary vasculature, which increases pulmonary arterial pressure.
It isn’t clear why some patients fail to resorb their blood clots, but risk factors for persistent clots include:
- Large pulmonary emboli
- Recurrent pulmonary emboli
- Insufficient anticoagulation
Group 5 – PH due to unclear multifactorial mechanisms
Group 5 PH is rare and beyond the scope of a non-specialist. Causes of Group 5 PH tend to be systemic pathologies affecting haematological or metabolic systems.
Clinical features
The clinical features of pulmonary hypertension are relatively non-specific.
Due to the non-specific nature of the symptoms of pulmonary hypertension, it means diagnosing the condition is challenging. Clinicians require a high index of suspicion. Indeed, one survey found that almost half of patients with PH had to see four doctors before they were diagnosed.
We can split the clinical features of pulmonary hypertension into three aspects:
- Initial non-specific respiratory symptoms
- Symptoms of right heart failure develop with disease progression.
- Symptoms that point towards the underlying cause or PH group.
1. Initial non-specific respiratory symptoms
- Progressive breathlessness
- Weakness
- Tiredness
2. Symptoms of right heart failure which develop with disease progression
- Peripheral oedema
- Ascites
- Angina (due to increased RV oxygen demand)
- Raised JVP
- Splitting of the second heart sound
- Right ventricular parasternal heave
3. Symptoms which point towards the underlying cause or PH group
These vary according to the underlying condition. For example, clubbing might point toward interstitial lung disease.
Investigations and diagnosis
The principal first-line investigation for the diagnosis of pulmonary hypertension is an echocardiogram.
Many specialist investigations are used to diagnose pulmonary hypertension, however, the most important first-line, non-invasive test is an echocardiogram.
Echocardiography
The first step in investigation is to confirm whether or not pulmonary hypertension is present. The first-line test for this is echocardiography. In the context of PH, echocardiography provides us with two specific measurements:
- Estimated pulmonary artery systolic pressure (ePASP)
- A raised ePASP increases the likelihood of PH
- Tricuspid regurgitant jet velocity (TRV)
- Increased pulmonary artery pressure and right ventricular remodeling frequently lead to tricuspid regurgitation.
- The higher the velocity of the regurgitant jet, the more likely pulmonary hypertension is to be present.
Echocardiography offers an additional useful measurement; we can use it to look at the left ventricle. If there are echocardiographic findings in keeping with PH and echocardiographic findings of left heart failure, this means a likely diagnosis is Group 2: PH due to Left Heart Disease.
For some patients with pulmonary hypertension, the diagnosis can be made based on history, examination, and echocardiography alone. In other patients, the diagnosis may be less clear. For example, echocardiography can sometimes be normal despite PH being present. If the history and examination are suspicious for PH, but the echocardiogram is normal, the next most appropriate investigation is right heart catheterisation.
Right heart catheterisation
During right heart catheterisation, a catheter is threaded through a vein into the right atrium, where a balloon is inflated. The inflated balloon ‘floats’ the catheter in the direction of blood flow i.e. into the right ventricle, and then on into a pulmonary artery. Once in the pulmonary artery, the catheter can be used to directly measure the pulmonary artery pressure. One important measurement is the pulmonary artery wedge pressure (PAWP), which reflects the pressure in the left atrium by indirectly measuring the pressure in the pulmonary capillary bed. The wedge pressure measurement helps figure out what’s causing the high pulmonary pressure. It shows whether the problem is because the left-sided heart failure (post-capillary) or if it’s due to issues in the blood vessels of the lungs themselves (pre-capillary).
Work-up
Patients presenting with the symptoms of PH (e.g. breathlessness or features of right-sided heart failure) are likely to undergo many other investigations as part of their workup.
The following results of these investigations point towards a diagnosis of PH:
- CXR: may be normal or show dilatation of the proximal pulmonary arteries, right atrium, or right ventricle.
- ECG: may be normal if no right-ventricular strain. ECG changes (e.g. right-axis deviation, P-pulmonale) are in keeping with right ventricular strain, right atrial enlargement, and right ventricular hypertrophy
Investigations to identify an underlying cause
As mentioned above, if echocardiography identifies both pulmonary hypertension and left heart failure, and the left heart failure is severe enough to explain pulmonary hypertension, then the patient can be categorised to Group 2: PH due to Left Heart Disease. If left-sided heart failure is not present, further tests will be required to identify the underlying cause of the PH as follows:
- Group 1: Screen for common causes (e.g. HIV, autoimmune testing)
- Group 2: See 'Echocardiography' above
- Group 3: Pulmonary function tests, high-resolution CT chest, consider sleep studies (e.g. obstructive sleep apnoea)
- Group 4: CT pulmonary angiography, ventilation/perfusion scanning (a chronic PE will show as an area/areas of mismatched ventilation and perfusion).
- Group 5: Usually consider when all other investigations unremarkable or clear alternative cause
Reversibility
Importantly, patients with Group 1 PH (i.e. PAH) should undergo testing for any reversibility.
Patients with Group 1 PH (i.e. PAH) should undergo acute vasoreactivity testing (AVT). AVT involves administering a calcium channel blocker, and assessing whether pulmonary hypertension improves in response to it (measured using a right heart catheter).
Roughly 20% of patients with Group 1 PH will be vasoreactive. Knowledge of whether or not a patient is vasoreactive (i.e. responds to a calcium channel blocker) will help guide management (see below).
Management
Management of pulmonary hypertension depends on the underlying cause.
Management of PH should also be led by pulmonary hypertension specialists. The management strategy depends on the underlying cause, which is why establishing the correct diagnosis is so important.
Group 1: Pulmonary arterial hypertension
The management of pulmonary arterial hypertension can broadly be divided into conservative, medical, and surgical:
- Conservative:
- Exercise as tolerated. This might be self-directed exercise, or via pulmonary rehabilitation exercise classes.
- Administer routine vaccinations.
- Avoid smoking.
- Maintain a normal body mass index.
- Counsel regarding pregnancy risk (30-50% mortality risk in presence of PH) and contraception if desired.
- Medical:
- Home oxygen therapy for patients with hypoxia
- Diuretics (e.g. furosemide) for symptoms of right heart failure (e.g. peripheral oedema, hepatic congestion)
- Vasoreactive patients can be trialled on a calcium channel blocker (e.g. nifedipine)
- Non-vasoreactive patients can be trialled on the following:
- Endothelin receptor antagonist (e.g. bosentan): Endothelin usually functions as a potent vasoconstrictor. As such, an endothelin antagonist reduces vasoconstriction.
- Phosphodiesterase 5 inhibitor (PDE5I) (e.g. sildenafnil): Under normal conditions, nitric oxide stimulates production of cyclic GMP to bring about its effects of vasodilation. Cyclic GMP (cGMP) is then broken down by phosphodiesterases, terminating nitric oxide’s vasodilatory effects. PDE5Is inhibit the breakdown of cGMP, therefore enhancing nitric oxide’s vasodilatory effects.
- Prostacyclin: Prostacyclin is a vasodilator via a cAMP pathway. Administering exogenous prostacyclin therefore encourages pulmonary vasodilation. Epoprostenol is the name of prostacyclin when used as a drug.
- Surgical
- Bilateral lung or heart-lung transplantation should be considered for patients with:
- Rapidly progressive disease
- Class III or IV WHO functional class
- Failure of medical therapy
- Bilateral lung or heart-lung transplantation should be considered for patients with:
Group 2: Left heart disease
Treatment is directed at improving left-sided heart disease and/or left-sided heart failure. Improved left-sided heart function should reduce the backpressure in the pulmonary circulation.
See our Note on Heart Failure for more details on the management of heart failure.
Group 3: Chronic lung disease and/or hypoxaemia
Treatment is mainly directed at the underlying lung disease.
Group 4: Pulmonary artery obstruction
Patients should start and/or continue anticoagulation, which prevents blood clots from growing. However, clot removal with pulmonary artery thromboendarterectomy (PTE) is the only potentially curative treatment for Group 4 PH. Patients need to be assessed for suitability for PTE, including answering the following questions:
- Is the patient well enough to tolerate major thoracic surgery?
- Which pulmonary arteries are affected?
PTE can only remove clots in proximal pulmonary arteries (i.e. main, lobar, or segmental pulmonary arteries). Clots in more distal pulmonary arteries are not usually accessible.
Group 5: Unclear of multifactorial
These conditions are rare and management is likely to be directed at the underlying cause.
Prognosis
The five-year mortality for group 1 pulmonary arterial hypertension is 50%.
Group 1 PAH is a progressive disease and the five-year mortality is estimated at around 50%. Once a patient has developed right heart failure secondary to PAH, median life expectancy falls to around 12 months. Mortality related to PAH is usually caused by right heart failure.
Conversely, the prognosis for groups 2-5 usually depends on the underlying cause, the severity of that illness, and its response to treatment. Taking groups 2 and 3 as an example, left heart failure or lung conditions severe enough to cause pulmonary hypertension and/or right heart failure suggest that the underlying condition is relatively serious.
Last updated: September 2024
References:
-
UpToDate: The epidemiology and pathogenesis of pulmonary arterial hypertension (Group 1).
-
UpToDate: Treatment and prognosis of pulmonary arterial hypertension in adults (Group 1).
-
Radiopedia. Sharma, R. Pulmonary Hypertension.
-
2013/14 NHS Standard Contract for Pulmonary Hypertension Centres
-
Pulmonary hypertension: a guide for GPs. Connolly, M.; Kovacs, G. British Journal of General Practice 2012; 62 (604): e795-e797. DOI: 10.3399/bjgp12X658467
-
An overview of the 6th World Symposium on Pulmonary Hypertension. Galiè, N.; McLaughlin V.; Rubin, L.; Simonneau, G. European Respiratory Journal Jan 2019, 53 (1) 1802148; DOI: 10.1183/13993003.02148-2018
-
The pathophysiology of chronic thromboembolic pulmonary hypertension. Simonneau, G.; Torbicki, A.; Dorfmüller, P.; Kim, N. European Respiratory Review Mar 2017, 26 (143) 160112; DOI: 10.1183/16000617.0112-2016
-
Pulmonary Hypertension Association UK. Appraisal of the drug treatments used in Pulmonary Arterial Hypertension (PH). Submission to the National Institute for Health & Clinical Excellence May 2007.
Have comments about these notes? Leave us feedback